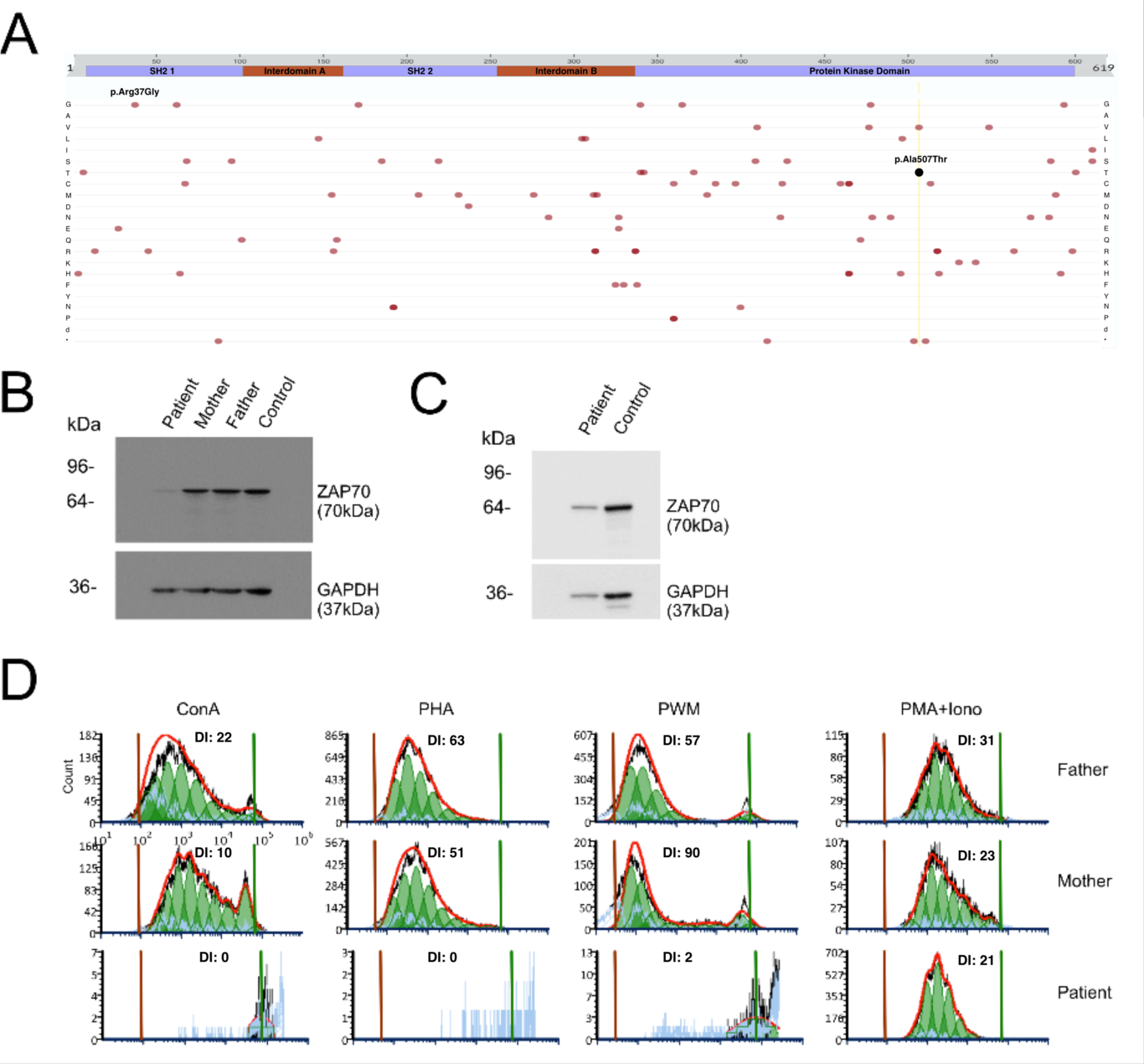
γδ T Cells and Novel Genetic Variants in ZAP70 Deficiency in An InfantIsma Shah, MDa; Samuel Chiang, PhDb,c; Li Yang, MD PhDb; Nagako Akeno, PhDb; Allison Kelly, DOa; Jason White, DOc; Emi Caywood, MDd; Sharon Hwang, MDa; and Trong Le, MDaaDivision of Allergy & Immunology, Nemours Children’s Hospital, Wilmington, DE, and Thomas Jefferson University, Philadelphia, PAbDivision of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OHcDepartment of Pediatrics, University of Cincinnati, Cincinnati, OHdDivision of Pediatric Hematology/Oncology, Nemours Children’s Hospital, Wilmington, DECorresponding Author Information:Name: Trong Le, MDMailing Address: 1600 Rockland Road, Wilmington DE 19703Telephone Number: 302-651-4321Fax Number: 302-651-5403Email address: [email protected] of Interest: NoneKeywords: ZAP70, γδ T Cells, pathogenic variants, newborn screening, NBS, SCID, PJP, pneumocystis jiroveciiTo The Editor:Clinical Implications:Early diagnosis and appropriate treatment of ZAP70 deficiency heavily depend on accurately classifying identified genetic variants. Reporting verified novel pathogenic variants and immunophenotypic features plays a crucial role in aiding the classification of genetic variants and improving patient outcomes.Most reported pathogenic variants in ZAP70 involve biallelic homozygous recessive or compound heterozygous loss-of-function mutations, resulting in abnormal thymic development, impaired TCR signaling in peripheral T cells, and loss of kinetic proofreading for ligand discrimination.1,2 These mutations cause various clinical phenotypes, including SCID, immune dysregulation, autoimmunity, malignancy, and late-onset combined immunodeficiency. ZAP70-related SCID is characterized by severe CD8+ lymphopenia and normal levels of dysfunctional CD4+ T cells, which are often missed due to TREC levels exceeding newborn screening cutoffs.3,4,5 Loss-of-function mutations in the kinase domain primarily contribute to the SCID phenotype (Fig. 1A).Herein we report a case of ZAP70 deficiency due to novel compound heterozygous mutations. Patient born at term to non-consanguineous parents with a normal SCID newborn screen and no history of severe infections or failure to thrive (weight 42% for age). At 7 months, she had acute hypoxemic ventilator-dependent respiratory failure. CBC showed Hb 10.4 g/dL, WBC 15900/µL (ANC 5900/µL, ALC 9700/µL), and platelets 115/µL. Respiratory viral panel (RVP) was negative. Urine culture had >1000,000 CFU/mL E. Coli, and blood cultures were negative. Further work-up revealed abnormal CXR and chest CT with contrast, demonstrating bilateral interstitial opacities, diffuse pneumonitis, and bronchioloalveolar lavage confirming Pneumocystis jirovecii pneumonia (PJP). IV trimethoprim/sulfamethoxazole (TMP/SMX) and prednisone were initiated. HIV testing was negative. Immune evaluation was notable for normal to elevated absolute lymphocyte counts, mildly decreased IgG (282 mg/dL), IgA (76 mg/dL), IgM (75 mg/dL), positive tetanus (0.05 IU/mL) and isohemagglutinin (32) titers, CD4+ cells, (4,742 cell/µL), severely reduced CD8+ cells (207 cells/µL), normal CD19+, and CD16+CD56+ cell counts. Inverted CD45RA+/CD45RO+ (1:6) ratio was observed. Maternal engraftment studies were negative. CD3+ proliferation studies revealed essentially absent responses to Concanavalin A (ConA; DI:0), phytohemagglutinin (PHA; DI:0), pokeweed mitogen (PWM; DI:2). Normal T-cell proliferation (DI:21) in response to mitogenic stimuli (phorbol-12-myristate-13-acetate [PMA] + ionomycin) bypassing TCR ligation6 (Fig. 1D) suggests intact response and abnormal TCR signaling defect. This indicates potential inherited mutations in the TCR-associated ZAP70 gene.The most striking immunophenotypic findings of ZAP70 combined immunodeficiency are normal levels of non-functional CD3+CD4+ T-cells and severely decreased levels of CD3+CD8+ T-cells with normal TCR Vβ repertoire.7 CD8+TCR heterodimer was assessed using flow cytometry to evaluate for TCR-α/β and TCR-γ/δ expression (Fig. 2A). In the control, the vast majority of circulating CD8+ TCR consists of the α/β heterodimer (99%), with less than 1% being γ/δ heterodimer. In the patient, TCR-γ/δ CD8+ lymphocytes are the predominant subset (63%), while α/β T lymphocytes make up a minor subset (36.7%). The ratio of surface γ/δ to α/β TCR expression has been suggested as a measure to diagnose T cell immunodeficiencies.8 Our patient’s CD8+ surface expression of γ/δ to α/β TCR has a characteristic ratio, which can serve as an immunophenotypic marker to distinguish ZAP70 deficiency from other selective CD8+ deficiencies related to genetic defects in TAP1, TAP2, TAPBP, B2M, and PIK3CG. In addition to the unique expression of γ/δ heterodimer on CD8+ T-cells, we observed a 3-fold increase in the expression of CCR6+CD8+ (10%) and CCR6+CD4+ (46.9%) T-cells compared to the control (Fig. 2B). The response to stimulation of IL-17A+ CCR6+CD4+and IL-2+CCR4+CD4+cells was similar in both the patient and the control, with no clear differences observed (Fig. 2C).Initially misclassified as unknown significance, the novel biallelic ZAP70 variants in our patient were likely pathogenic based on further laboratory findings. Western blot analysis showed no ZAP70 expression, and T cell proliferation was absent in response to TCR-mediated stimuli (Fig. 1B, 1D). The paternally-inherited variant, Exon 3.c.109C>G (p.Arg37Gly), affects the N-SH2 domain and impacts phosphotyrosine binding by substituting Arginine with Glycine. The maternally-inherited compound heterozygous sequence variant, c.1519G>A, causes the amino acid substitution p.Ala507Thr in the protein kinase domain. This variant (Fig. 1A) has not been reported as pathogenic in ZAP70-related CID cases to our knowledge. Our patient tested negative for other candidate gene variants, micro deletion and duplication syndromes, aneuploidy, and triploidy.Affected children rarely survive beyond their second year without hematopoietic stem cell transplantation (HSCT). Our patient received an HSCT from her haploidentical father after recovering from the acute illness. The reduced-intensity conditioning regimen for the HSCT included alpha/beta T cell depletion and several drugs, including anti-thymoglobulin, thiotepa, methylprednisolone, busulfan, and fludarabine. The patient engrafted on Day +13 and tolerated the conditioning, but on Day +16, she exhibited symptoms of sinusoidal obstructive syndrome (SOS), which was treated with defibrotide and supportive care. The patient was discharged on Day +44. Post-transplant, the patient received prophylactic treatment and monthly immunoglobulin replacement therapy. Immune evaluation showed normal reconstitution, with normal mitogen stimulation and high immune cell response. Two years later, the patient experienced mixed chimerism, but continued to show evidence of immune reconstitution without requiring additional therapy.ZAP70 deficiency is a rare combined immunodeficiency disorder with normal levels of dysfunctional CD4+ T cells. It is often missed due to high TREC levels that exceed newborn screening cutoffs. Timely diagnosis at initial presentation relies on reported pathogenic variants and unique immunophenotypic features. We have identified a novel pathogenic variant and a potential unique immunophenotype of a polarized γδ CD8+ T-cell subset in a patient with ZAP70 deficiency, which could aid in future diagnosis and improved outcomes.ReferencesSharifinejad, N., et al., Clinical, Immunological, and Genetic Features in 49 Patients With ZAP-70 Deficiency: A Systematic Review. Front Immunol, 2020. 11: p. 831.Swamy, M., ZAP70 holds the key to kinetic proofreading for TCR ligand discrimination. Nat Immunol, 2022. 23(9): p. 1293-1294.Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM. Defective T cell receptor signaling and CD8++ thymic selection in humans lacking zap-70 kinase. Cell. 1994;76:947–58.Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, Grunebaum E. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130:177–83.Jilkina, O., et al., Retrospective TREC testing of newborns with Severe Combined Immunodeficiency and other primary immunodeficiency diseases. Mol Genet Metab Rep, 2014. 1: p. 324-333.Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, Parslow TG. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science. 1994;264:1596–9.Roifman CM, Dadi H, Somech R, Nahum A, Sharfe N. Characterization of zeta-associated protein, 70 kd (ZAP70)-deficient human lymphocytes. J Allergy Clin Immunol. 2010;126:1226–33.e1.Garcillan, B., et al., gammadelta T Lymphocytes in the Diagnosis of Human T Cell Receptor Immunodeficiencies. Front Immunol, 2015. 6: p. 20.