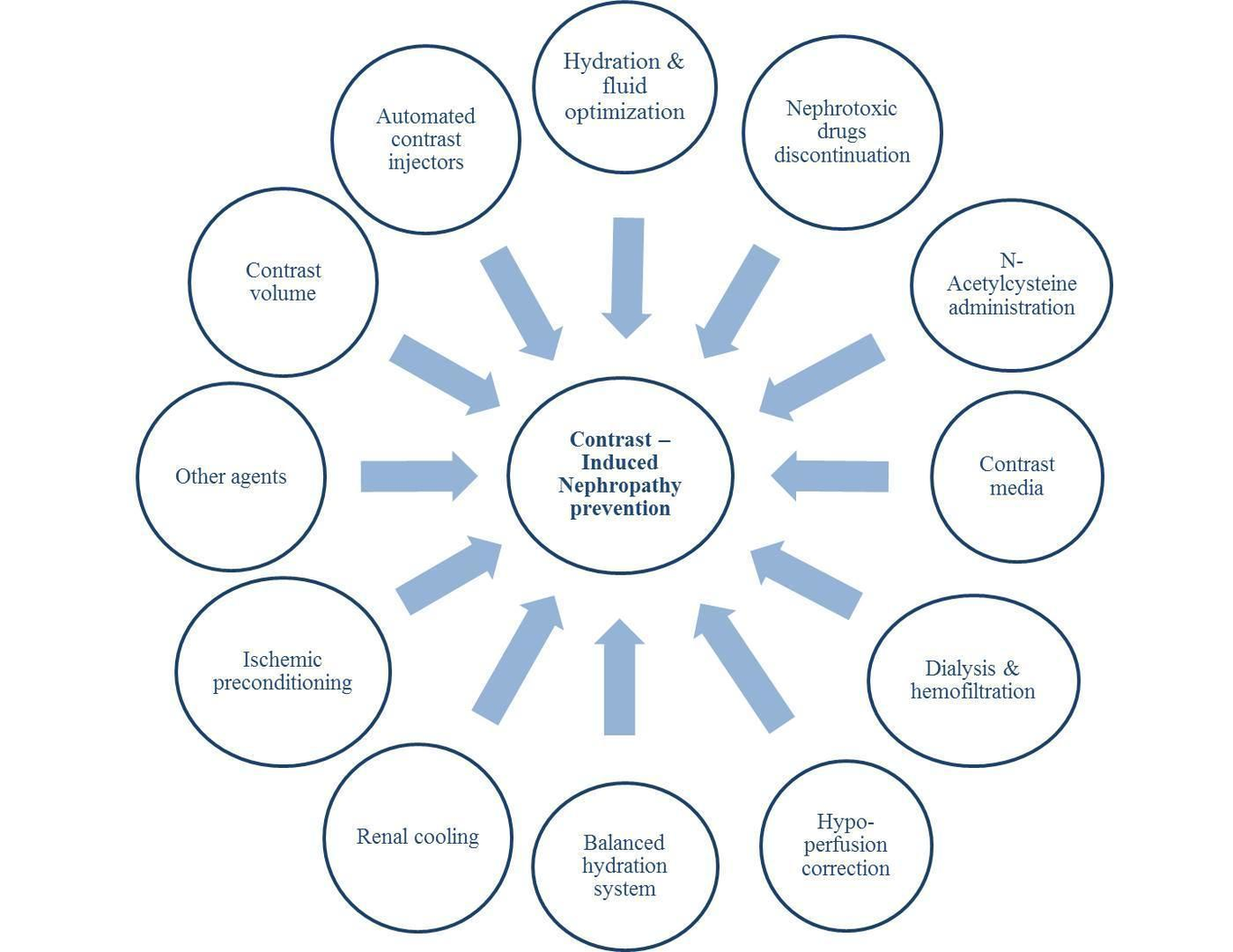
Introduction:Epidemiology:Each year, there are millions of procedures done in the United States that utilize iodinated contrast agents. The main risk of iodinated contrast is kidney injury which could lead to morbidity and mortality (1–6). Contrast-induced nephropathy (CIN), also known as contrast-induced acute kidney injury (CI-AKI), is one of the most common causes of impairment of renal function in the United States (7–10) and is the third common cause of hospital-acquired renal insufficiency (11). Different studies have used various definitions for contrast-induced nephropathy including an increase in serum creatinine ≥0.5 mg/dl or ≥25% from baseline creatinine within 24 to 72 hours after contrast medium administration (3,6,12–16). The mortality rate is increased among patients who developed CIN during and after hospitalization, especially among those who required dialysis (6,17,18). Therefore, any preventive measures that can reduce CIN risk can be lifesaving with a reduction in mortality and morbidity in patients undergoing iodinated contrast exposure.Brief Summary of CIN preventive measures:● Hydration and fluid optimizationSeveral methods have been proposed to prevent CIN in clinical settings. However, none of them has been proved to be consistently effective except for hydration and reduction in the amount of contrast exposure. Hydration is the most common prophylactic technique to reduce CIN occurrence in a way that all high-risk patients undergoing contrast exposure should receive appropriate hydration if possible before the procedure in high-risk patients and after the procedure in all patients if feasible without contraindication to hydration (14,19–23). It has been shown that intravenous fluid administration with isotonic saline is more effective compared to the half saline infusion (14,22). However, the optimal fluid volume and infusion rate is controversial. Current guidelines recommend intravenous administration of 1-1.5 ml/kg/h of normal saline six hours before and after contrast injection (24). With respect to proper fluid administration, left ventricular end-diastolic pressure (LVEDP) can be assessed and adjusted accordingly. This theory was assessed in the POSEIDON trial. Their findings suggested patients who received adjusted fluid based on LVEDP had a significantly lower risk of CIN after cardiac catheterization (relative risk: 0.41, 95% confidence interval (CI): 0.22 – 0.79, P= 0.005). However, three cases of shortness of breath, probably in the context of pulmonary edema, were reported in both intervention and control groups (25). Also, Maioliet et al. used bioimpedance vector analysis (BIVA) for the assessment of body fluid status. After randomization of low BIVA patients to normal or double volume normal saline administration, they found no significant difference in CIN occurrence defined by standard criteria (increase serum creatinine by ≥ 0.3 mg/dl within 48 hours) between those groups (10.8% vs. 4.7%, P= 0.08, respectively) (26).● Avoidance of nephrotoxic agentsAnother factor that can raise CIN risk might be related to nephrotoxic drugs. Although there are not enough trials to strongly prove the benefit of nephrotoxic drugs discontinuation before contrast exposure, it is generally recommended to hold potentially nephrotoxic drugs including nonsteroidal anti-inflammatory drugs, aminoglycosides, vancomycin, sulfonamides, penicillins, amphotericin, loop diuretics, and metformin in high-risk patients. The latter drug has been associated with metabolic acidosis which might predispose kidneys to the development of CIN but this concept has not been proven (27,28).● N-Acetylcysteine administrationN-Acetylcysteine (N-AC) was initially reported by Tepel et al. to be protective against contrast-induced nephropathy in a small trial (29). However, numerous trials and meta-analyses have completely failed to show any benefit and therefore its use is not recommended (29–31).● Type of contrast mediaContrast media typed based on osmolality is thought to be important for CIN pathogenesis and has been categorized into three different types based on the osmolality (high osmolar, low osmolar, and iso-osmolar) (32). Initially, several studies have shown that iso-osmolar contrast media have the lowest risk of CIN incidence in comparison to low-osmolar contrast agents (33–36), but numerous other trials failed to show any significant differences in the occurrence of contrast-induced nephropathy (37–40).● Dialysis and hemofiltrationIn terms of dialysis and hemofiltration which directly removes the contrast from the systemic circulation, there is no clinical evidence suggesting prophylactic use of dialysis can prevent CIN (41). No benefit has been reported for post-procedural dialysis either (42). Marenzi et al. reported the use of hemofiltration might be beneficial in the prevention of CIN (43). However, it remains unclear whether it was related to increased clearance through dialysis or due to alkalinizing agents used during filtration.● Treatment of hypoperfusionDue to the negative effect of renal hypoperfusion, regardless of its etiology, with contrast administration resulting in increased CIN risk, utilization of short time assisted devices increasing cardiac output might reduce this risk. Flaherty and colleagues performed a randomized clinical trial and found usage of a Microaxial percutaneous assist device (Impella) was associated with a lower likelihood of acute kidney injury among high risk percutaneous coronary intervention (PCI) patients with reduced left ventricular ejection fraction ≤ 35% (odds ratio (OR): 0.13, 95% CI: 0.09 – 0.31, P< 0.001) (44). These findings might be associated with resultant reduced CIN risk. However, larger studies are warranted.● Balanced hydration systemAnother proposed mechanism in CIN prevention has been attributed to a balanced hydration procedure. This process has been suggested based on the theory that as urine output becomes higher, the contrast concentration in kidneys would become lower ultimately resulting in decreasing CIN risk. Briguori et al. implemented Renal Insufficiency After Contrast Media Administration Trial II (REMEDIAL II) trial to assess the feasibility of the RenalGuard system (PLC Medical Systems, Inc, Franklin, MA) in the prevention of CIN. Briefly, the mentioned system consists of closed-loop fluid management that consistently monitors and evaluates hydration status and urine output. 294 candidates for coronary or peripheral angiography/angioplasty with an estimated glomerular filtration rate (eGFR) of ≤ 30 ml/min/1.73 m2 and/or risk score of at least 11 were selected and randomly allocated to control (sodium bicarbonate and N-AC administration) or RenalGuard (hydration with saline and N-AC under RenalGuard system control with furosemide administration) group. The intervention group received an initial bolus for 30 minutes and furosemide (0.25 mg/kg) would be prescribed to increase urine output to ≥ 300 ml/h. They found CIN was significantly decreased in the RenalGuard arm compared to controls (11% (16 out of 146 subjects) vs. 20.5% (30 out of 146 subjects), OR: 0.47, 95% CI: 0.24 – 0.92). Different administration routes of N-AC (oral agent for controls and intravenous route for intervention group) resulting in probable variable bioavailability of the drug as well as their reported data applicable to a subset of chronic kidney disease (CKD) patients might be considered for extension of the outcomes (45).Likewise, the Induced Diuresis With Matched Hydration Compared to Standard Hydration for Contrast-Induced Nephropathy Prevention (MYTHOS) trial using the RenalGuard system was performed for CKD patients who underwent coronary procedures. 170 subjects with eGFR< 60 ml/min/1.73 m2 were randomly assigned to standard intravenous saline hydration as a control group (n= 83) or furosemide with matched hydration as an intervention group (n= 87). The intervention arm received 250 ml of normal saline as well as 0.5 mg/kg of furosemide to reach a urine output of more than 300 ml/h. Patients in the intervention group experienced CIN less frequently rather than controls (4.6% vs. 18%, P= 0.005). Single-center and not-blinded study design, as well as pre-determined hydration protocol in the intervention group, were some limitations related to the mentioned project (46).● Renal coolingAlso, a cooling renal method based on the theory of decreasing oxidative injury in lower temperatures in the context of contrast injection has been announced. However, it did not show any promising outcome in terms of CIN prevention. For instance, Stone and colleagues performed a randomized trial and allocated 128 cardiac catheterization candidates with CKD (estimated creatinine clearance: 20-50 ml/min) to control (n= 70) and intervention (n= 58) groups. In addition to hydration, the latter group underwent systemic hypothermia at 33-34 °C starting before contrast injection toward three hours post-procedure followed by rewarming to 36 °C with a rate of 1 °C per hour afterward. CIN was observed in 18.6% and 22.4% of normothermia and hypothermia groups, respectively. However, there was no significant association neither in unadjusted nor in adjusted models (OR: 1.27, 95% CI: 0.53 – 3.00, P= 0.59 and OR: 0.83, 95% CI: 0.18 – 3.78, P= 0.81, respectively) (47).● Ischemic preconditioningThe hypothesis of ischemic preconditioning, as multiple short cycles of ischemia and reperfusion in one organ, could be effective on another organ, on reduction of CIN has been tested in a randomized clinical trial on 100 subjects which revealed four 5-minute inflation-deflation cycles of blood pressure cuff to 50 mmHg above each patient systolic blood pressure before coronary angiography (CA) had been associated with a decreased likelihood of CIN compared to controls (OR: 0.21, 95% CI: 0.07 – 0.57, P= 0.002) (48). Although this procedure can be applied in all clinical settings, further studies with a larger sample size are required.● Other agentsOne small study showed infusion of sodium bicarbonate might be more effective in the prevention of CIN rather than isotonic saline (49). However, subsequent larger trials failed to prove this association (25,26). Therefore, sodium bicarbonate is not recommended to be used for this purpose by the Consensus Working Panel (22).Other pharmacologic agents include ascorbic acid, diuretics, mannitol, calcium channel blockers, fenoldopam, dopamine, atrial natriuretic peptide, L-arginine, theophylline, and statins have been reported in the literature in terms of CIN prevention with controversial results (50–62).The role of contrast volume● Contrast volume as a risk for CINContrast volume has been shown to be an independent risk factor for CIN (63–65). It has been previously proved the amount of contrast correlates with the incidence of CIN (66). After a data analysis of 53780 vascular interventions, Lee et al. indicated CIN was correlated with CKD stage in a way that the incidence of AKI in the context of contrast administration raised with each CKD stage (CKD stage 1: 0.39%, CKD stage 2: 0.45%, CKD stage 3: 1.5%, CKD stage 4: 4.3% and CKD stage 5: 7.5%). They suggested the risk of post-contrast AKI could be reduced by using safe thresholds of contrast volume (67).Rihal et al.’s study reported the volume of contrast media administered during the PCI was correlated with acute renal failure (6). Kooiman and colleagues analyzed data from 82,120 PCI procedures and found patients who received high contrast, as defined by division of contrast volume over calculated creatinine clearance resulting in more than 3, had increased CIN odds in both univariate and multivariate regression models (OR: 1.61, 95% CI: 1.46 – 1.79, P< 0.001 and OR: 1.77, 95% CI: 1.58 – 1.98, P< 0.001, respectively) (68). Likewise, another observational study on 561 patients suffering from myocardial infarction who underwent PCI revealed CIN was significantly higher among those with a contrast ratio (measured by administered contrast volume divided by calculated maximum contrast agent dose) of more than 1 in comparison to the ratio of less than one (34.6% vs. 3%, P< 0.001) (65). Kane et al. reported the rate of CIN in patients with CKD undergoing CA could be reduced by ultra-low contrast volumes (69). However, even small amounts of contrast can deteriorate renal function, especially among high-risk patients (70). A small study on 30 patients with eGFR< 45 ml/min/1.73m2 underwent CA/PCI with ultra-low volume contrast media showed utilization of this kind of agent was safe with no reported increased serum creatinine 48 hours post-procedure (71). However, a single study design and small sample size are potential limitations needed to be considered. 123 subjects with at least stage 3 of CKD experienced CA/PCI was selected by Kelly and colleagues. They used a novel ultra-low contrast delivery technique with an automated contrast injector for their procedures and reported a CIN rate of 3.3%. Quite a small sample size, as well as retrospective study design and performance in a single-center, should be considered for the generalization of their findings (72). Although the CIN rate was lower among CKD patients who underwent PCI with ultra-low contrast (n= 8) compared with the conventional group (n= 103) in another retrospective study, the difference was not statistically significant (0 vs. 15.5%, P= 0.28). Asymmetric sample distribution between groups and their small cohort size might limit their outcomes (73).Mariani et al. proposed the theory of zero contrast volume and performed MOZART (Minimizing cOntrast utiliZation With IVUS Guidance in coRonary angioplasTy) trial to assess whether intravascular ultrasound (IVUS) could decrease contrast exposure compared to the routine method. 83 PCI candidate patients were selected and randomly assigned to routine angiography (n= 42) or IVUS method (n= 41) with matched clinical and laboratory data. The median contrast volume was significantly lower in IVUS rather than in the routine angiography group (20 ml, interquartile range (IQR): 12.5 – 30 ml vs. 64.5 ml, IQR: 42.8 – 97 ml, P< 0.001). Also, the ratio of contrast volume to creatinine clearance was remarkably lower in the IVUS group (0.4, IQR: 0.2 – 0.6 vs. 1.0, IQR: 0.6 – 1.9, P< 0.001) (74). Although they found a promising outcome, the higher cost of IVUS might be a limiting factor for usage in clinical settings. On the other hand, it has been suggested that contrast volume reduction before contrast exposure may lower the risk of CIN (75).● Methods for reducing contrast volume administrationIn terms of reducing contrast volume administration, few studies are available. Mehran et al. performed a randomized clinical trial to assess the efficiency of contrast reduction in patients with underlying renal diseases who underwent CA. 578 patients in stage III (eGFR between 30 and 60 ml/min) and IV (eGFR between 20 and 30 ml/min) of CKD with at least two further criteria of New York heart association (NYHA) functional class of III or IV of heart failure, diabetes mellitus (treated with either insulin or oral agents), anemia, hypertension, albuminuria or age of at least 75 years were randomly assigned to hydration (n= 286) or hydration plus AVERT system group (n= 292). The latter system is a contrast modulation system designed to adjust the pressure of contrast injection toward the patient. The relative reduction in contrast volume was 15.5% (hydration group: 101.3 ± 71.1 ml vs. hydration plus AVERT group: 85.6 ± 50.5 ml, P= 0.02). The distribution of AKI induced by contrast did not differ significantly (26.6% vs. 27%, P= 0.70, respectively) (76). Likewise, Gurm and colleagues performed an observational study on 114 patients with eGFR of 20 – 60 ml/min/1.73m2 to assess the feasibility of contrast volume reduction during CA or PCI using DyeVertTM Plus Contrast Reduction System (DyeVert Plus System, Osprey Medical). Data analysis of 105 successfully recruited patients revealed the contrast volume saving of 40.1 ± 8.8% per each performed procedure. AKI induced by contrast agent was observed in three (2.6%) of patients (77). The small sample size and observational design of the study should be considered for the generalization of reported data.● Automated contrast injection devicesAlthough data analysis of 60,884 candidates who underwent PCI revealed contrast agent usage was lower in centers that used automated contrast injectors compared to those centers not used this method (199 ± 84 ml vs. 204 ± 82 ml, P< 0.0001), no difference had been found in terms of CIN occurrence (3.11% vs. 3.42%, P= 0.15) (78). On the other hand, Minsinger and colleagues performed a meta-analysis and found automated contrast injectors decreased contrast volume up to 45 ml per subject (95% CI: 0.78 – 0.93, P< 0.001), and it was associated with a 15% reduction in CIN compared to manual injection methods (OR: 0.85, 95% CI: 0.78 – 0.93, P< 0.001) (79).