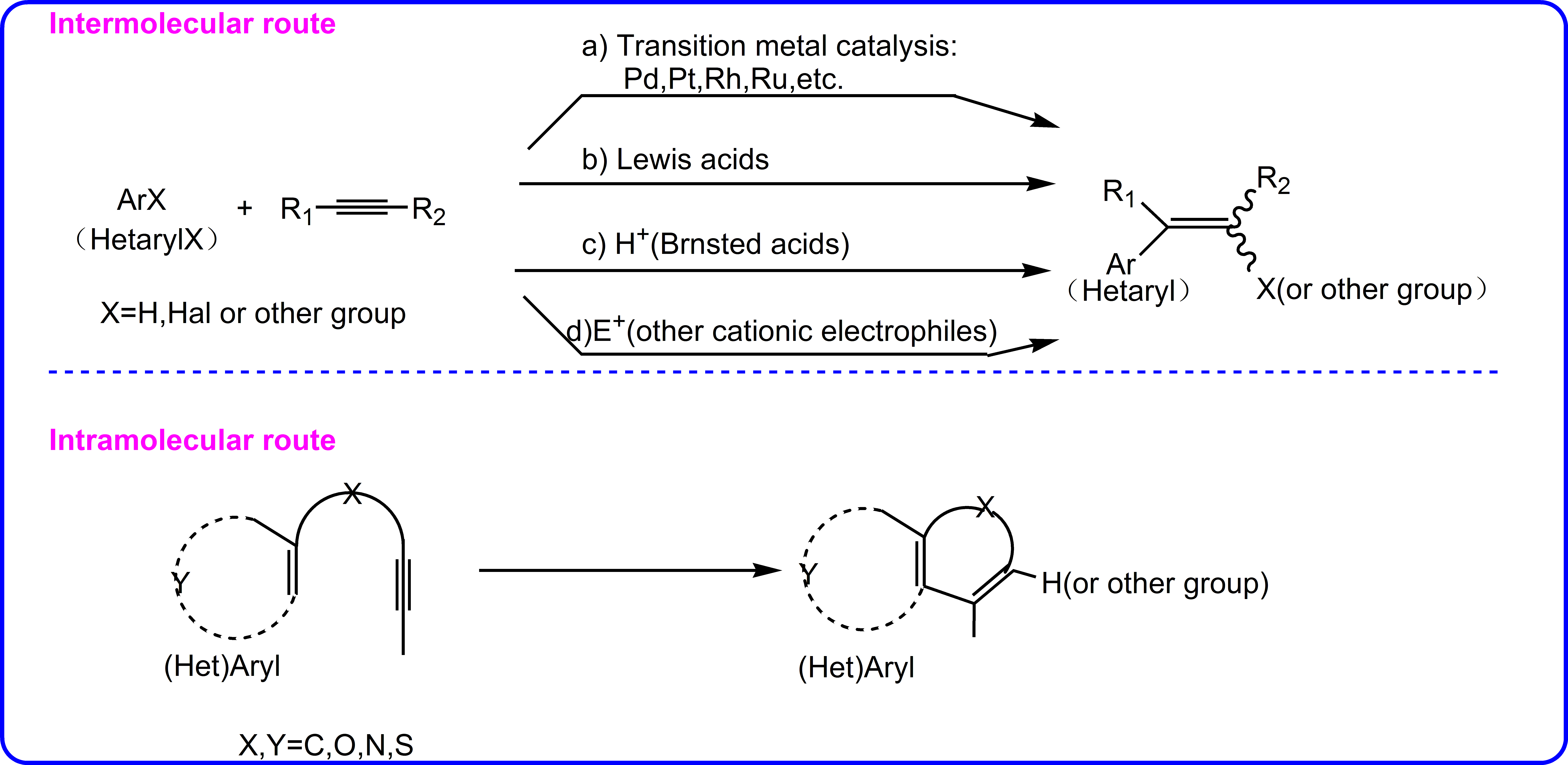
The ligand-promoted palladium-catalyzed hydroarylation of alkynes with arenes without directing group is able to furnish alkenyl chlorides via a 1,4-chlorine migration or trisubstituted alkenes. This reaction is challenging due to bidentate N, N ligand and electron-neutral arenes have rarely been reported to afford good yields. We carried out density functional theory calculations to better understand the elementary steps of the reaction and unveil the ligand effects and origin of substituent-controlled chemoselectivity of challenging C-H activation. For the n-propyl-substituted substrate, CMD process is the rate-determining step of the catalytic reaction. And the chemoselectivity is controlled by oxidative addition with the C-Cl bond cleavage and protonation process. However, for the reaction with 3,5-dimethylphenyl-substituented substrate, the key step of the whole catalytic cycle is the protonation process. The stronger electrostatic attractions, repulsive force and aryl substituent effects result in reverse chemoselectivity. Bidentate ligand L1 (2-OH-1,10-phenanthroline) reacts with Pd(OAc)2 to form a most stable square-planer species, which is different from the one formed by ligand L2(1,10-phenanthroline). The steric repulsion are found to be mainly responsible for no product with L2 as the ligand, which is different from as proviously reported.