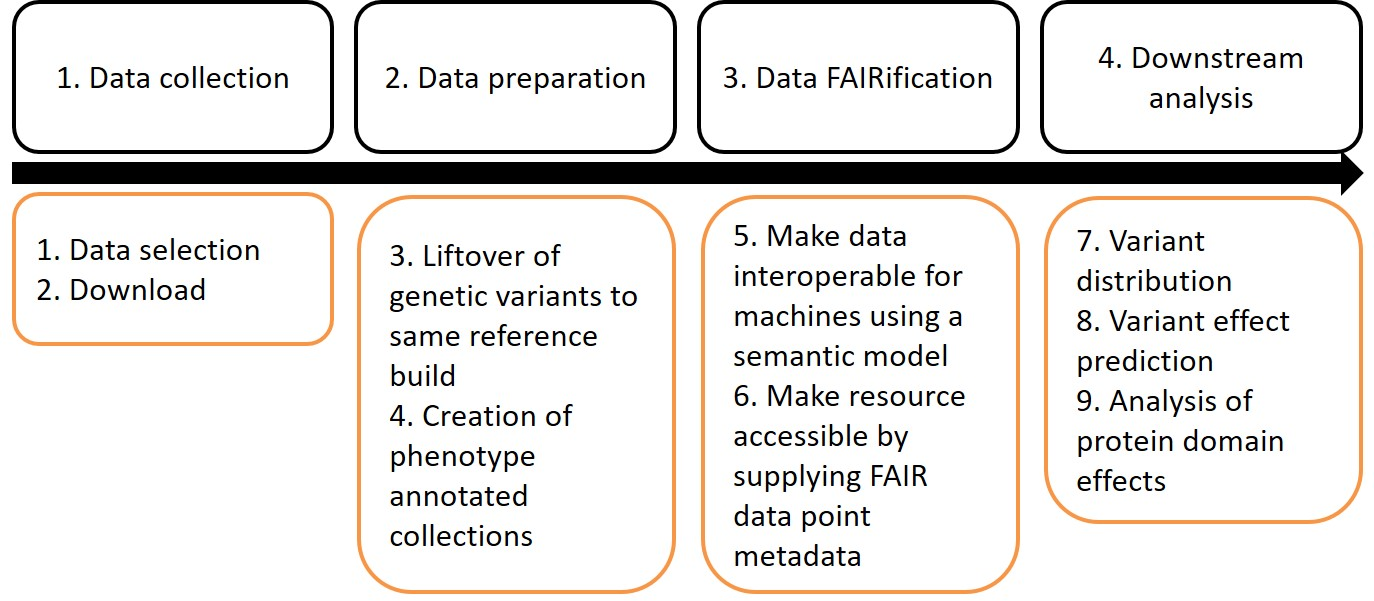
Rett syndrome (RTT) is a rare neurological disorder mostly caused by a genetic variation in MECP2. Various RTT causing and benign variants in MECP2 have been identified and due to the advent of sequencing in clinical diagnosis new variants are identified daily. Making new MECP2 variants and the related phenotypes available provides data for better understanding of disease mechanisms and faster identification of variants for diagnosis. This is, however, currently hampered by the lack of interoperability between genotype-phenotype databases. Here, we demonstrate on the example of MECP2 in RTT that by making the genotype-phenotype data more Findable, Accessible, Interoperable, and Reusable (FAIR), we can facilitate prioritization and analysis of variants. In total, 10,968 MECP2 variants were successfully integrated. Among these variants 863 unique confirmed RTT causing and 209 unique confirmed benign variants were found. This dataset was used for comparison of pathogenicity predicting tools, protein consequences, and identification of ambiguous variants. Prediction tools generally recognised the RTT causing and benign variants, however, there was a broad range of overlap. 19 variants were identified, which were annotated as both, disease-causing and benign suggesting that there are more disease-causing factors than a mutation contributing to the disease development.