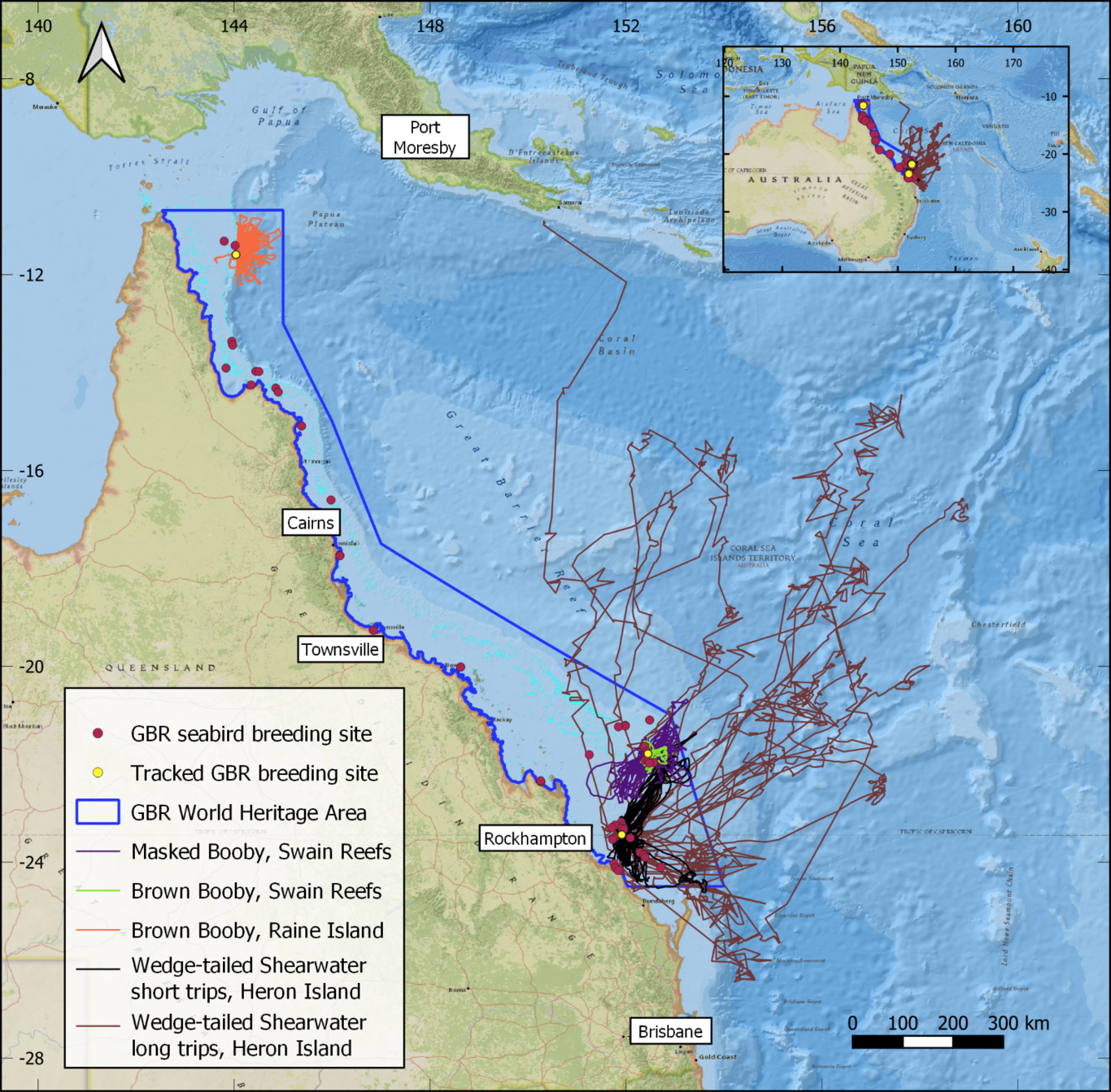
Conservation of breeding seabirds typically requires detailed data on where they feed at sea. Ecological niche models (ENMs) can fill data gaps, but rarely perform well when transferred to new regions. Alternatively, the foraging radius approach simply encircles the sea surrounding a breeding seabird colony (a foraging circle), but overestimates foraging habitat. Here, we investigate whether ENMs can transfer (predict) foraging niches of breeding tropical seabirds between global colonies, and whether ENMs can refine foraging circles. We collate a large global dataset of tropical seabird tracks (12000 trips, 16 species, 60 colonies) to build a comprehensive summary of tropical seabird foraging ranges and to train ENMs. We interrogate ENM transferability and assess the confidence with which unsuitable habitat predicted by ENMs can be excluded from within foraging circles. We apply this refinement framework to the Great Barrier Reef (GBR), Australia to identify a network of candidate marine protected areas (MPAs) for seabirds. We found little ability to generalise and transfer breeding tropical seabird foraging niches across all colonies for any species (mean AUC: 0.56, range 0.4-0.82). Low global transferability was partially explained by colony clusters that predicted well internally but other colony clusters poorly. After refinement with ENMs, foraging circles still contained 89% of known foraging areas from tracking data, providing confidence that important foraging habitat was not erroneously excluded by greater refinement from high transferability ENMs nor minor refinement from low transferability ENMs. Foraging radii estimated the total foraging area of the GBR breeding seabird community as 2,941,000 km2, which was refined by excluding between 197,000 km2 and 1,826,000 km2 of unsuitable foraging habitat. ENMs trained on local GBR tracking achieved superior refinement over globally trained models, demonstrating the value of local tracking. Our framework demonstrates an effective method to delineate candidate MPAs for breeding seabirds in data-poor regions.