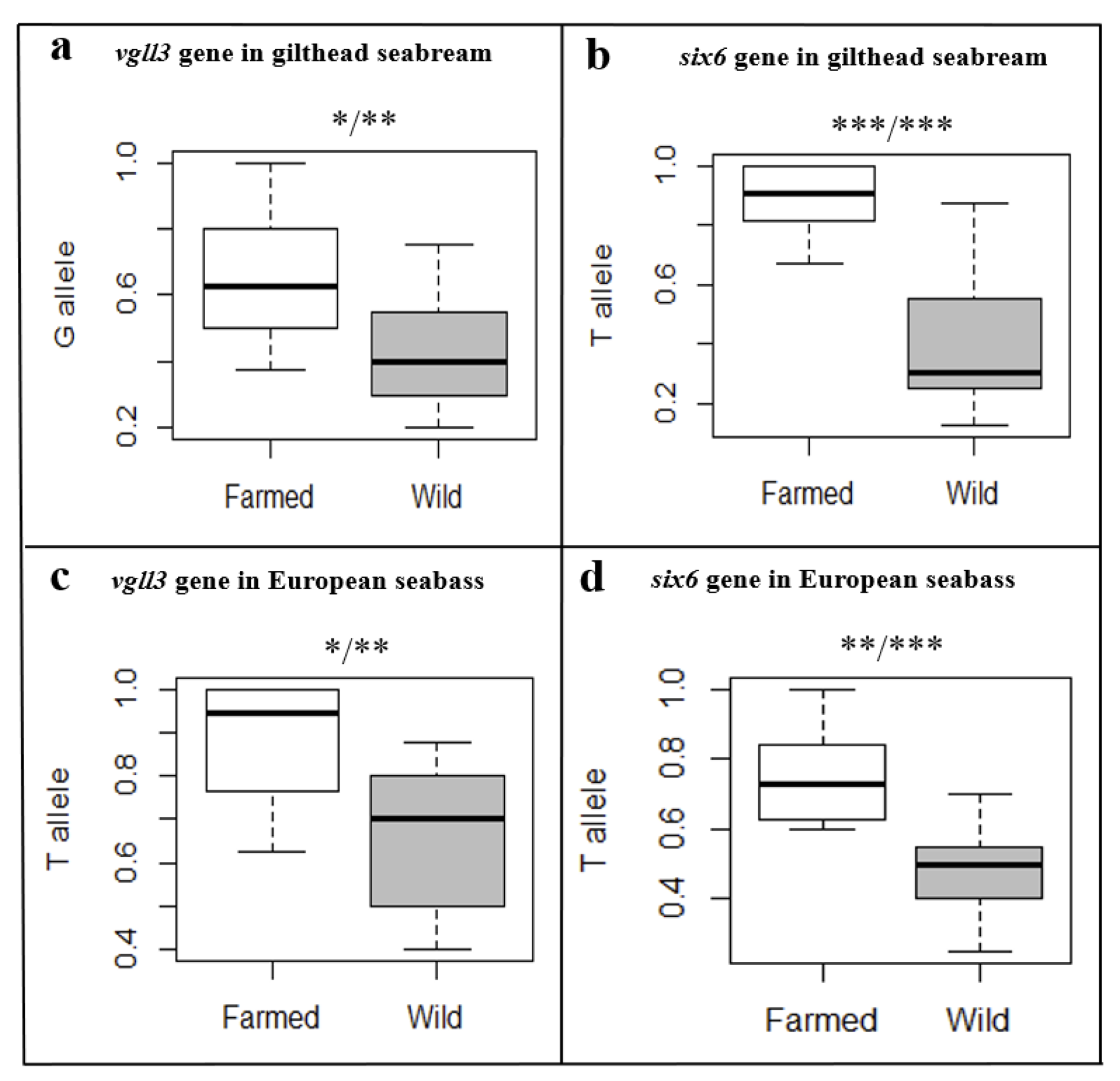
Genome scans offer a comprehensive method to explore genome-wide variation associated with traits under study. However, connecting individual genes to broader functional groupings and pathways is often challenging, yet crucial for understanding the evolutionary mechanisms underlying these traits. This task is particularly relevant for multi-trait processes such as domestication, which are influenced by complex interactions between numerous genetic and non-genetic factors, including epigenetic regulation. As various traits within the broader spectrum of domestication are selected in concert over time, this process offers an opportunity to identify broader functional overlaps and understand the integrated genetic architecture underlying these traits. In this study, we analyzed approximately 600,000 SNPs from a Pool-Seq experiment comparing eight natural-origin and 12 farmed populations of European seabass in the Mediterranean Sea region. We implemented two genome scan approaches and focused on genomic regions supported by both methods, resulting in the identification of 96 candidate genes, including nine CpG islands, highlighting potential epigenetic influences. Many of these genes and CpG islands are in linkage groups previously associated with domestication-related traits. The most significantly overrepresented molecular function was ‘oxidoreductase activity’. Furthermore, a dense network of interactions was identified, connecting 22 of the candidate genes. Within this network, the most significantly enriched pathways and central genes were involved in ‘chromatin organization’, highlighting another potential epigenetic mechanism. Altogether, our findings underscore the utility of interactome-assisted pathway analysis in elucidating the genomic architecture of polygenic traits and suggest that epigenetic regulation may play a crucial role in the domestication of European seabass.