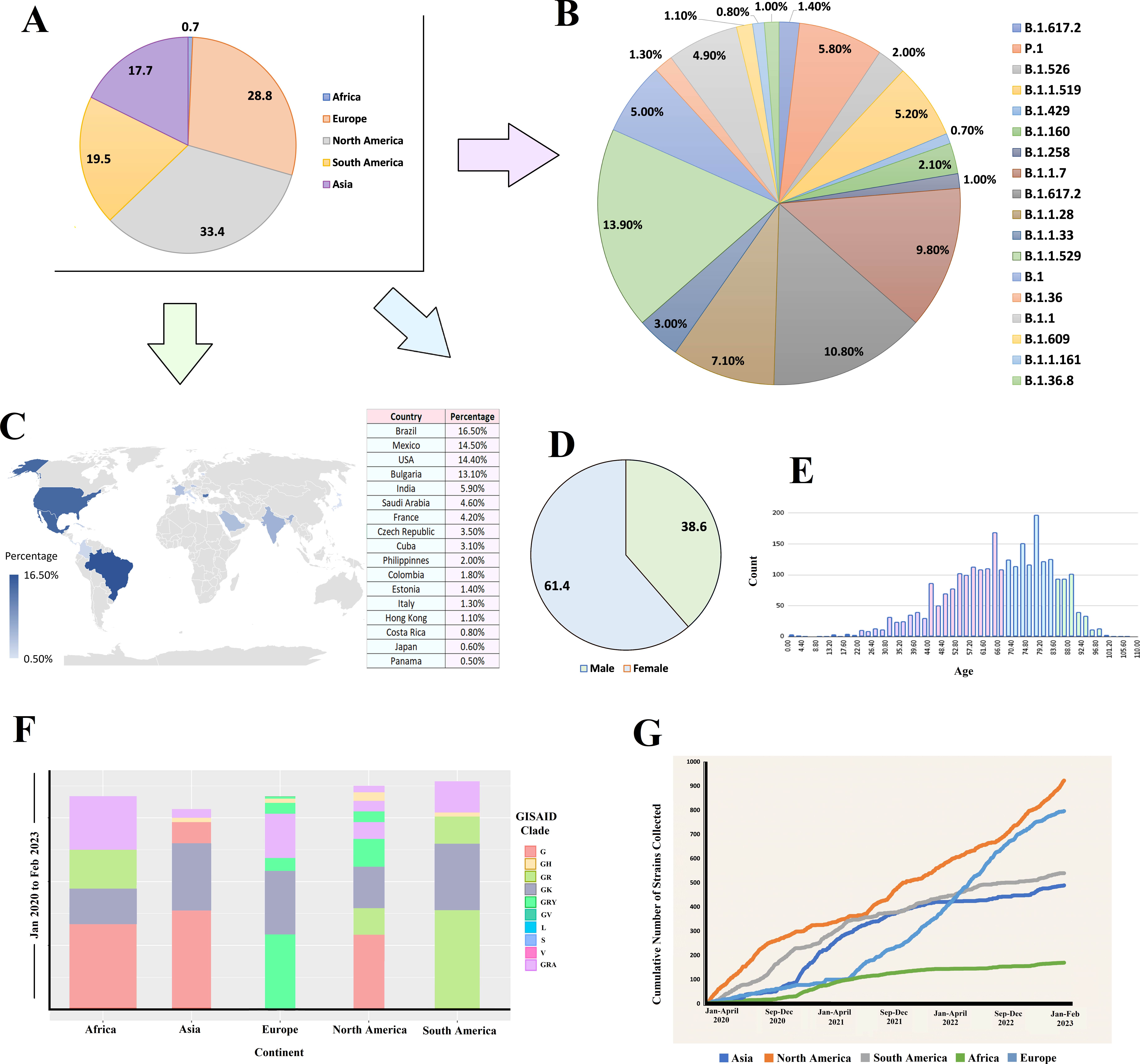
The identification of deleterious mutations in different variants of SARS-CoV-2, and their roles in the morbidity of COVID-19 patients are yet to be explored. Analyzing 5,724 complete genomes of SARS-CoV-2, sequenced from deceased COVID-19 patients globally during January 2020 to February 2023, we found that SARS-CoV-2 genomes of the deceased belonged to 21 Nextstrain clades, of which clade 20I (Alpha variant) was the most predominating clade followed by clade 20H (Beta variant) and clade 20J (Gamma variant). The highest percentage (33.4%) of SARS-CoV-2 genomes from deceased patients were sequenced from North America, while the lowest (0.98%) was from Africa. The “G” clade was found to be predominated in the SARS-CoV-2 genomes Asian, African, and North American regions whereas “GRY” clade outweighed in Europe. We identified 35,799 nucleotide mutations throughout the genome keeping the highest (n = 11,402) frequency in the spike protein. More importantly, 4,150 point-specific amino acids (aa) mutations in SARS-CoV-2 genomes, and of them, D614G (20%) and N501Y (14%) deleterious mutations in the spike protein were found as the top two mutations worldwide. We also detected five frequent deleterious aa mutations such as G18V, W45S, I33T, P30L, and Q418H, responsible for defining each clade of the SARS-CoV-2. Our novel findings could therefore be useful for genomic surveillance and monitoring the integrated pattern of SARS-CoV-2 infection, its emerging variants, and their impacts on developing effective vaccination and control methodology.