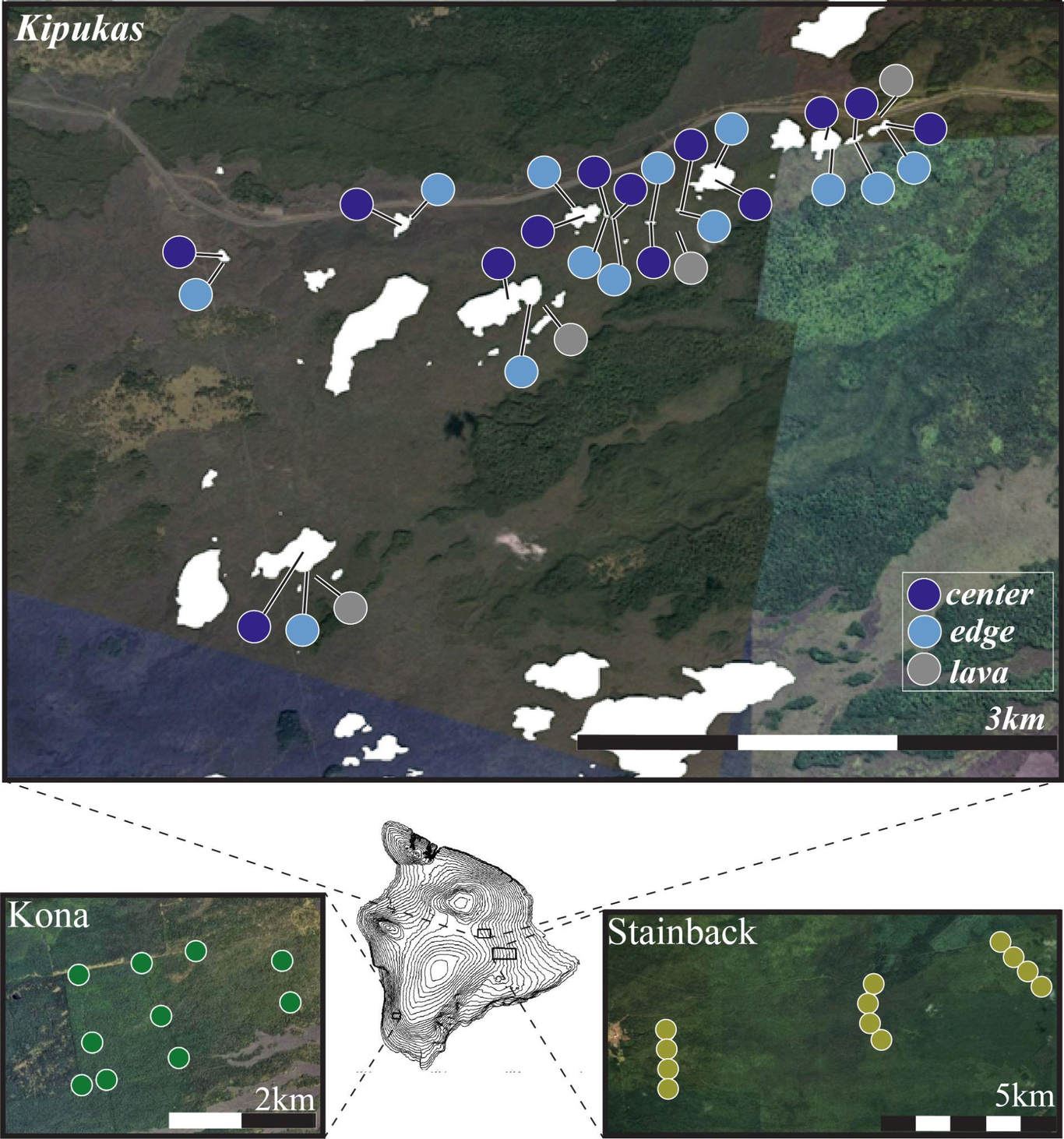
The term ‘habitat fragmentation’ is frequently associated with the biologically-destructive activities of human development, but an important evolutionary hypothesis posits that much of the biodiversity we see today was generated by episodic, natural habitat fragmentation. This hypothesis suggests that fragmentation can serve as a ‘crucible of evolution’ through the amplifying feedbacks of colonization, extinction, adaptation, and speciation. Interrogating the generality of this hypothesis requires measuring the repercussions of fragmentation at intra- and interspecific levels across entire communities. We use DNA metabarcoding to capture these repercussions from the scales of intraspecific differentiation to community composition in a megadiverse, ecologically foundational group, arthropods, using a natural habitat fragmentation experiment on patches of wet forest isolated by contemporary Hawaiian lava flows (kīpuka). We find a pronounced effect of area in kīpuka cores, where the taxonomic richness supported by a kīpuka scales with its size. Kīpuka cores exhibit higher intra- and interspecific turnover over space than continuous forest. Additionally, open lava, kīpuka edges, and the cores of small kīpuka (which are essentially entirely “edge”) host lower richness, are more biologically homogeneous, and have higher proportions of non-native taxa than kīpuka cores. Our work shows how habitat fragmentation isolates entire communities of habitat specialists, paving the way for genetic differentiation. Parsing the extent to which differentiation in kīpuka is driven by local adaptation versus drift provides a promising future avenue for understanding how fragmentation, and the different isolated communities created through this process, may lead to speciation in this system.