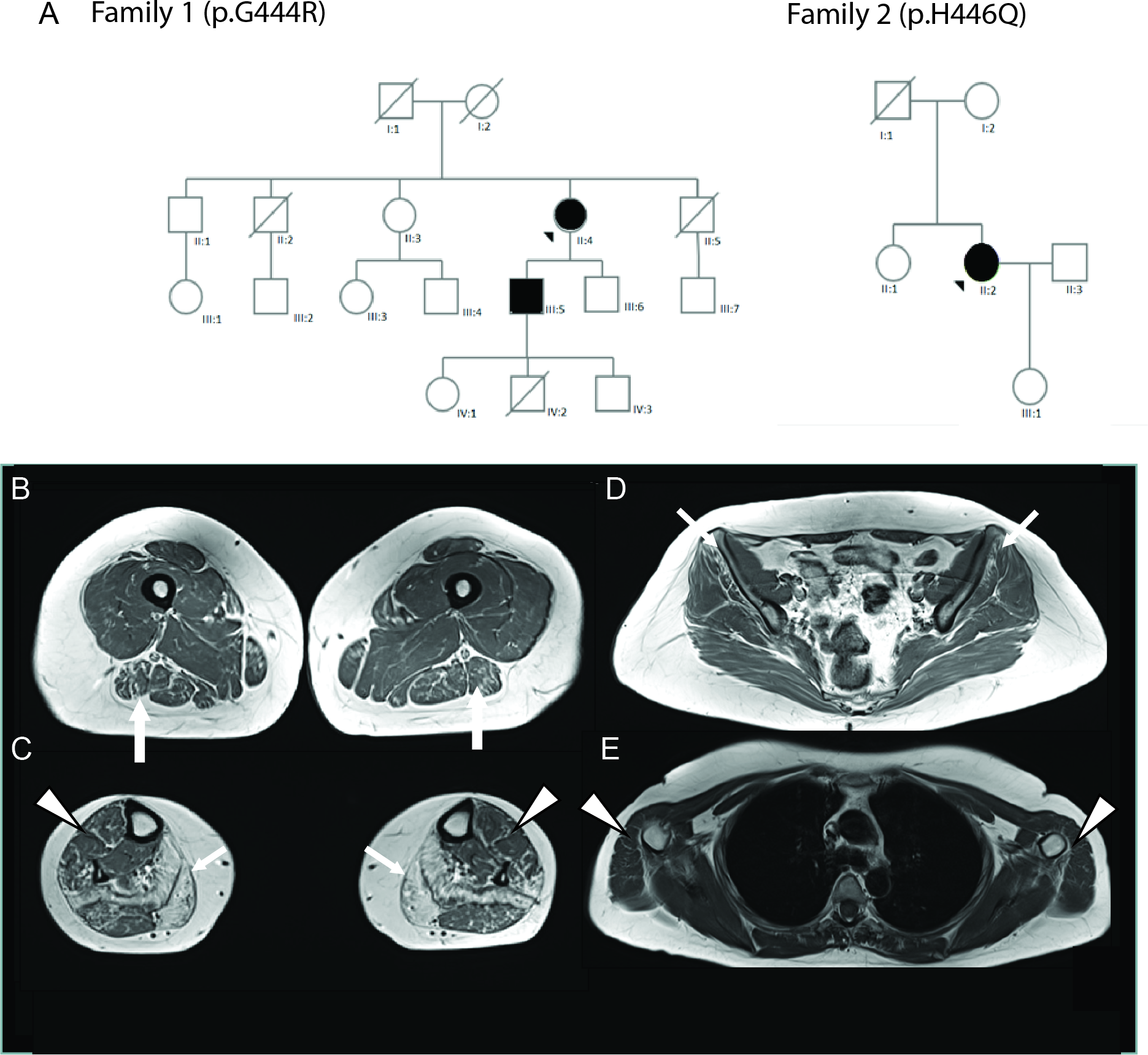
MORC2 gene encodes a ubiquitously expressed nuclear protein involved in chromatin remodeling, DNA repair, and transcriptional regulation. Heterozygous mutations in MORC2 gene have been associated with a spectrum of disorders affecting the peripheral nervous system such as Charcot-Marie-Tooth (CMT2Z), spinal muscular atrophy-like (SMA-like) with or without cerebellar involvement, and a developmental syndrome associated with impaired growth, craniofacial dysmorphism and axonal neuropathy (DIGFAN syndrome). Such variability in clinical manifestations associated with the increasing number of variants of unknown significance detected by next-generation sequencing constitutes a serious diagnostic challenge. Here we report the characterization of an in vitro model to evaluate the pathogenicity of variants of unknown significance based on MORC2 overexpression in a neuroblastoma cell line SH-EP or in cortical neurons. Likewise, we show that MORC2 mutants affect survival and trigger apoptosis over time in SH-EP cell line. Furthermore, overexpression in primary cortical neurons increases apoptotic cell death and decreases neurite outgrowth. Altogether, these approaches establish the pathogenicity of two new variants p.G444R and p.H446Q in three patients from two families. These new mutations in MORC2 gene are associated with autosomal dominant CMT and with adult late onset SMA-like phenotype, further increasing the spectrum of clinical manifestations associated with MORC2 mutations.