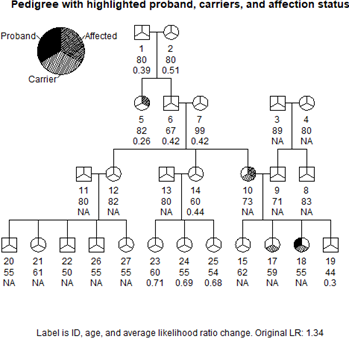
Clinical genetic sequencing tests often identify variants of uncertain significance (VUS). One source of data that can help classify the pothogenicity of variants is familial cosegregation analysis. Identifying and genotyping relatives for cosegregation analysis can be time consuming and costly. We propose an algorithm that describes a single measure of expected variant information gain from genotyping a single additional relative in a family. Then we explore the performance of this algorithm by comparing actual recruitment strategies used in 35 families who had pursued cosegregation analysis with synthetic pedigrees of possible testing outcomes if the families had pursued an optimized testing strategy instead. For each actual and synthetic pedigree, we calculated the likelihood ratio of pathogenicity as each successive test was added to the pedigree. We analyzed the differences in cosegregation likelihood ratio over time resulting from actual versus optimized testing approaches. Employing the testing strategy indicated by the algorithm would have led to maximal information more rapidly in 30 of the 35 pedigrees (86%). Many clinical and research laboratories are involved in targeted cosegregation analysis. The algorithm we present can facilitate a data driven approach to optimal relative recruitment and genotyping for cosegregation analysis and more efficient variant classification.