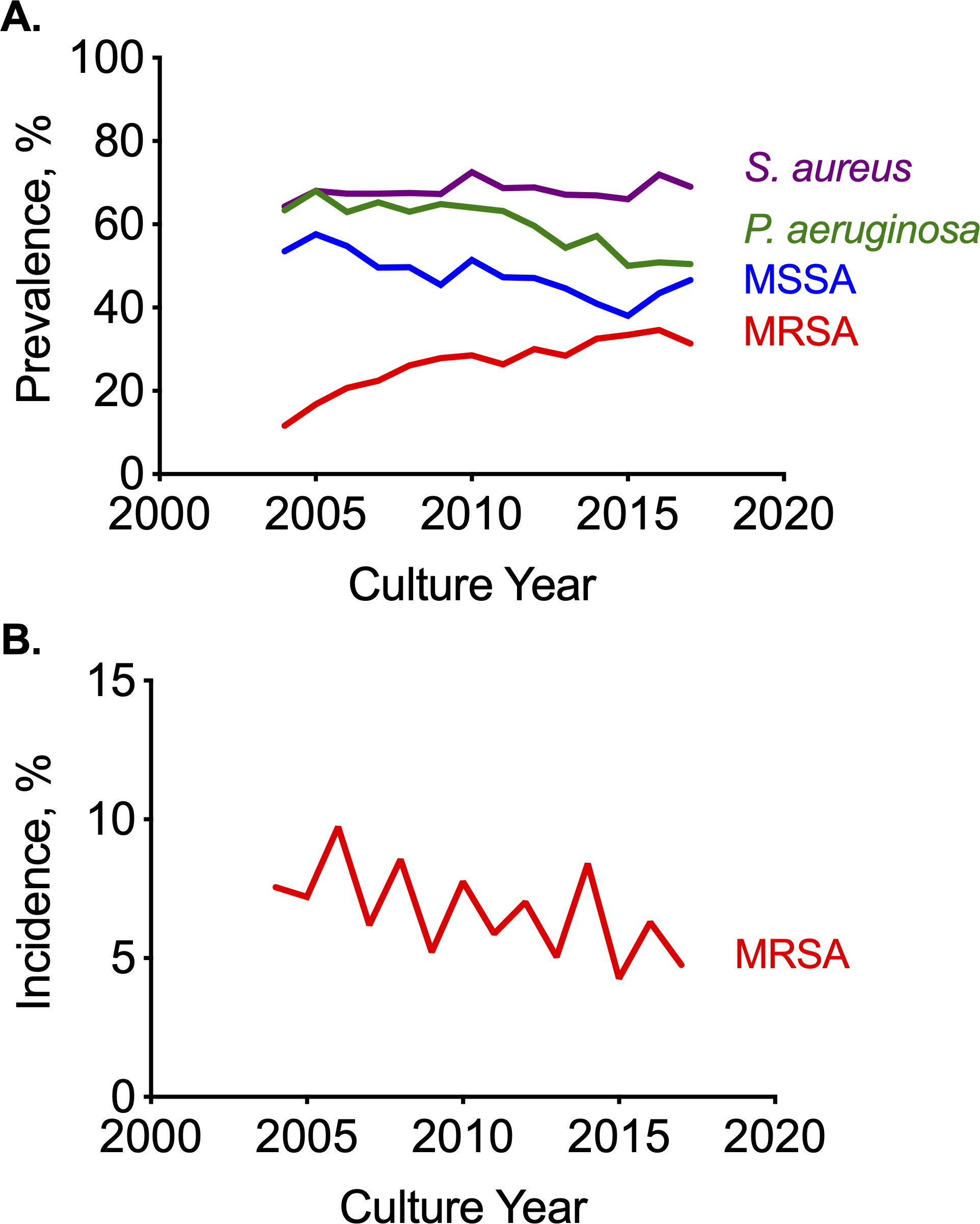
Rationale: Methicillin resistant Staphylococcus aureus (MRSA) is prevalent and consequential in cystic fibrosis (CF). Whole genome sequencing (WGS) could reveal genomic differences in MRSA associated with poorer outcomes or detect MRSA transmission. Objectives: To identify MRSA genes associated with low lung function and identify potential MRSA transmission in CF. Methods: We collected 97 MRSA isolates from 74 individuals with CF from 2017 and performed short-read WGS. We determined sequence type (ST) and the phylogenetic relationship between isolates. We aligned accessory genes from 25 reference genomes to genome assemblies. We classified the MRSA by accessory gene content and correlated the accessory genome to clinical outcomes. Results: The most prevalent ST were ST5 (N=55), ST105 (N=14), and ST8 (N=14). Closely related MRSA strains were shared by family members with CF, but rarely between unrelated individuals. Three distinct clusters of MRSA were identified by accessory genome content. The first included ST5 and ST105 strains and was common among older patients with lower FEV1. The second cluster included ST8, which was generally identified in younger patients. Sputum density of MRSA and Pseudomonas aeruginosa was higher in cultures from patients with ST5/ST105 compared to patients with ST8 at similar ages. Conclusions: In this CF cohort, we identified MRSA subtypes that predominate at different ages and differ by accessory gene content. ST5 and ST105 represented the most prevalent cluster of MRSA. ST8 MRSA was more common in younger patients and thus has the potential to rise in prevalence as these patients age.