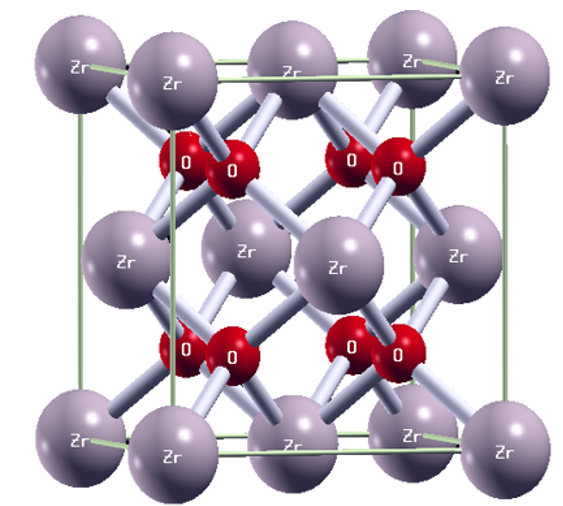
In this paper, we use the density functional theory (DFT) calculations under Quantum Espresso package to characterize the doping effect of sulfur substituting on the Zirconium dioxide ZrO2. Through the density of states and the band structure calculations, a direct band gap is appearing for the pure and doped studied system. The electronic properties analysis shows that the doping with sulfur could considerably decrease the band gap of doped ZrO2 by the presence of an impurity state of sulfur 3 p on the up spin of the valence band. The results of the ab-initio density functional theory investigations show that the substitutional sulfur dopants incorporated into the Zirconium dioxide ZrO2 drastically and affect the electronic structure of the studied material. In fact, the doping of Zirconium dioxide ZrO2 with appropriate concentration values of sulfur leads to band gap values in the interval (1-2) eV. We recall that the band structure and density of states can improve among others: the energy gap of this doped ZrO2 material. In fact, we have started from 1.3 eV for the pure ZrO2 to reach 1.2 eV for 9% of sulfur doping. This last energy gap value is suitable for photovoltaic application.