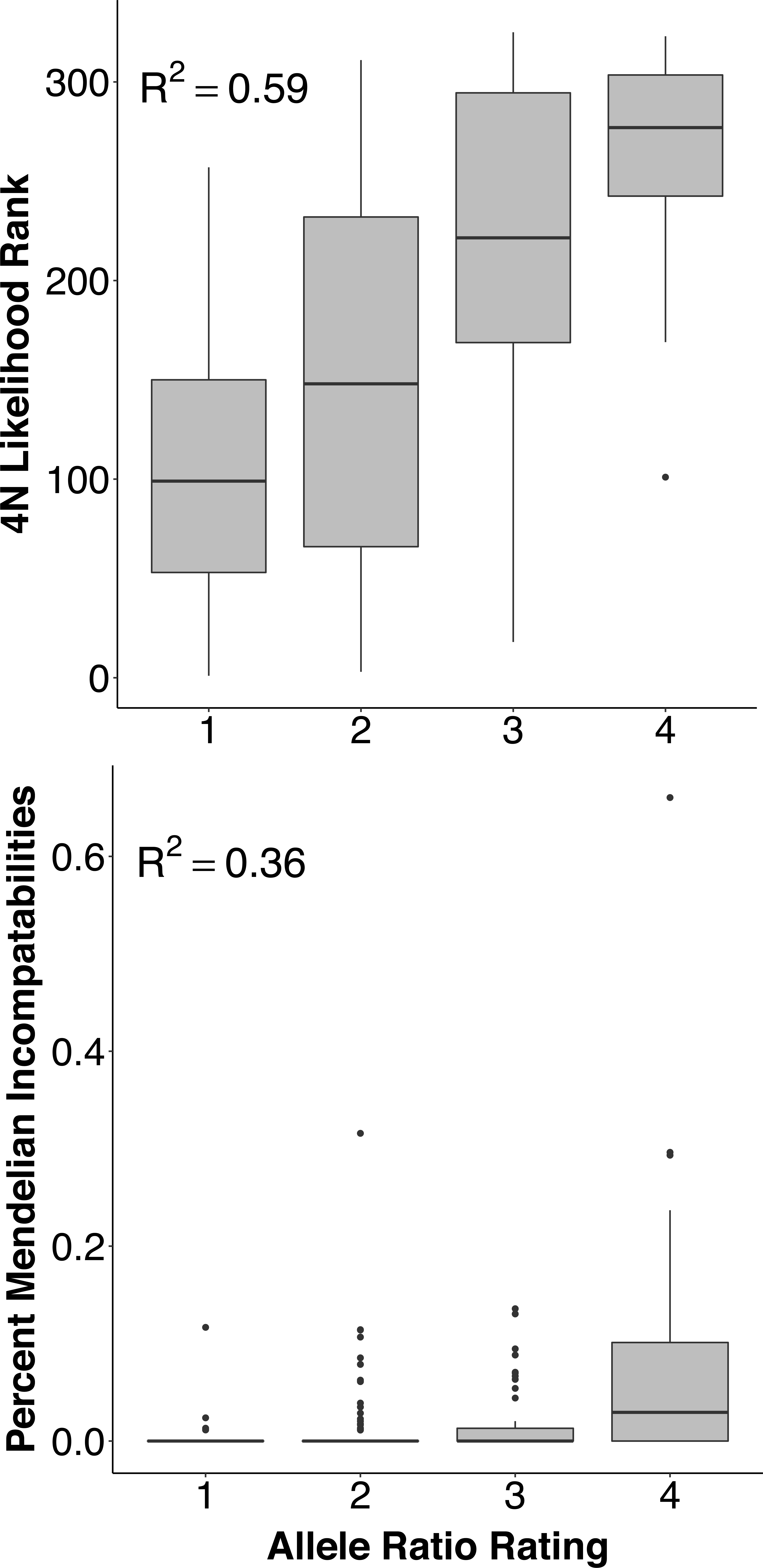
Polyploidization has played a critical role in the evolution of several major organism groups, including vertebrates, but much of our knowledge of the evolution of polyploids comes from allopolyploid and often rediploidized lineages, which partly reflects the difficulty of obtaining genotype data from polysomic genomes. We combined several contemporary methods to develop markers for single nucleotide polymorphisms compatible with simultaneous ploidy estimation and high throughput genotyping, and analyzed these data with recent software developments that accept polysomic data. We demonstrate the utility of this combination to develop genetic resources for polysomic species by applying it to the ploidy-variable and polysomic white sturgeon (Acipenser transmontanus), an imperiled species under conservation management in the Pacific Northwest. We introduce a primer and probe set for 325 SNP markers for use with the ‘Genotyping-by-thousands’ (GT-seq) method, and provide updated scripts that incorporate a function to estimate ploidy from each individual using read count data. We examine the reliability of tetrasomic inheritance in a large sample of paleo-octoploid individuals and the expected Mendelian inheritance patterns in known cross families. We then demonstrate our ability to use these data to infer parentage, relatedness, and other population genetic parameters. Our combined process thus improves the accessibility of genetic information to facilitate future investigations of white sturgeon and is expected to be widely applicable to other polyploid species.